
Relación de la Obesidad con la Diabetes Tipo 2
- Noticiero Medico
- 1 nov 2019
- 7 Min. de lectura

Dra. Magali Macías
La obesidad se considera un factor de riesgo para desarrollar Diabetes Mellitus tipo2. El aumento del tejido adiposo se ha relacionado con el aumento de la producción de citoquinas pro-inflamatorias, que junto a los ácidos grasos, parecen ser los responsables del desarrollo de la resistencia a la insulina.¹
Definición de Resistencia Insulínica (RI)
La insulina es una hormona anabólica secretada por las células beta del páncreas en respuesta a diversos estímulos, siendo la glucosa el más relevante. Su principal función es mantener la homeostasis glucémica y de otros sustratos energéticos. De esta forma, posterior a cada comida, la insulina suprime la liberación de ácidos grasos libres mientras que favorece la síntesis de triglicéridos en el tejido adiposo. Por otra parte, la insulina inhibe la producción hepática de glucosa, mientras que promueve la captación de glucosa por el tejido muscular esquelético y adiposo. En un estado de RI, la acción de esta hormona a nivel celular está reducida, lo que aumenta la secreción de insulina por el páncreas.²
Inicialmente, la hipersecreción de insulina mantiene la glucemia bajo control. No obstante, esta situación progresivamente lleva al denominado fracaso pancreático, cuando las células beta no sólo no son capaces de mantener la hipersecreción de insulina, sino que empiezan a deteriorarse disminuyendo la secreción de insulina. Este es el punto en que se diagnostica la mayoría de los casos de diabetes mellitus tipo 2 y síndrome metabólico.¹
Aunque la etiología de la RI todavía no está claramente establecida, se considera que existe un componente genético poligénico sobre el que actuarían factores ambientales: sedentarismo y comidas procesadas.³
Por otra parte, existen bases genéticas relacionadas con el desarrollo de la diabetes y obesidad, cabría señalar que durante los últimos años, los estudios de asociación del genoma completo (GWAS) han tenido un éxito sin precedentes al identificar locus que están involucrados en enfermedades comunes tales como la obesidad y la diabetes. Por ejemplo, existen más de 35 locus susceptibles que se han identificado para la diabetes tipo 2 y 32 para la obesidad. En el año 2006, el factor de transcripción Transcription factor 7-like 2 (TCF7L2) fue implicado en la diabetes tipo 2.¹
Señalización normal de la Insulina
La resistencia a la insulina puede desarrollarse por alguna anomalía en la cascada de señalización de insulina. La señalización normal de la insulina se produce a través de la activación de un receptor específico para la insulina, que pertenece a una subfamilia de receptores tirosina quinasas13. A diferencia de la mayoría de los receptores tirosina quinasa, el receptor activado de insulina directamente fosforila en múltiples residuos de tirosina el substrato del receptor de insulina (IRS)1. A pesar de que actualmente se han descrito cuatro miembros en la familia de IRS que están involucrados en la señalización de insulina, IRS-1 y 2 son los más importantes en el transporte de glucosa.¹
El siguiente nivel de señalización de insulina involucra la fosfoinositol-3 quinasa (PI3quinasa). La PI3 quinasa es un elemento clave en la respuesta metabólica de la insulina, que regula el transporte de glucosa, el efecto antilipolítico, la síntesis de ácidos grasos y la síntesis de glucógeno. La proteína quinasa B (PKB/Akt) pertenece a un nivel inferior en la regulación de la señalización de la insulina. Existen tres isoformas diferentes de esta enzima, pero sólo Akt2 parece ser la isoforma que media la sensibilidad a insulina en el músculo esquelético y en el hígado. El efecto final de la insulina es facilitar el transporte de glucosa que se realiza a través del transportador de glucosa 4 (Glut 4).³
Resistencia insulínica mediada por inflamación
La obesidad ha sido asociada a un estado inflamatorio crónico leve a moderado, que se manifiesta a nivel sistémico por un aumento de citoquinas y leucocitos circulantes. A nivel tisular y particularmente en el tejido adiposo, se caracteriza por infiltración de células inmunes. A nivel molecular, diversos tipos celulares (adipocitos, células endoteliales, leucocitos, células hepáticas, célula beta pancreática, neuronas, entre otras) manifiestan una mayor unión de factores de transcripción pro-inflamatorios (por ejemplo el factor nuclear kappa Beta o NFκB) a elementos de respuesta nuclear.²
En condiciones pro-inflamatorias, los mediadores inflamatorios se unen a los receptores de las membranas celulares, lo cual desencadena la migración del factor de transcripción NFкB desde el citosol al núcleo para la síntesis de nuevos mediadores inflamatorios. En estado basal, este factor de transcripción está inactivo en el citosol, unido a su inhibidor IкB, lo que le impide migrar al núcleo. En respuesta a una señal externa pro-inflamatoria (ej. TNFá), la proteína IKK induce la degradación de IкB, dejando a NFкB libre para migrar al núcleo, transmitiendo así la señal inflamatoria. Sin embargo, la proteína IKK también fosforila al sustrato del receptor de insulina 1 (IRS1). En condiciones fisiológicas, IRS1 se activa cuando está fosforilado en residuos de tirosina; sin embargo, la fosforilación de IKK ocurre en su residuo serina. Como consecuencia, hay una inhibición de la transducción de la señal insulínica, determinando una menor translocación del transportador de glucosa 4 (GLUT4) desde el citosol a la membrana celular, disminuyendo así la captación de la glucosa sanguínea (Figura 1). Como respuesta compensatoria, ocurre una hipersecreción de insulina, lo cual explica la típica hiperinsulinemia de los individuos con RI. De esta manera, una célula expuesta a un entorno inflamatorio es una célula resistente a insulina.²

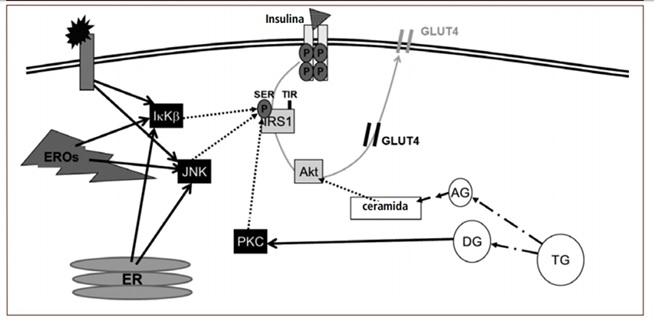
Figura 1. Eventos celulares que interfieren en la señalización insulínica
La insulina (triángulo) se une a su receptor de membrana, gatillando la fosforilación del mismo receptor y proteínas post-receptor (ej. sustrato del receptor de insulina 1 [IRS1], Akt, etc.). Esto determina la migración del transportador de glucosa 4 (GLUT4) a la membrana, lo que facilita la captación de glucosa. En un proceso inflamatorio, los mediadores inflamatorios se unen a su receptor de membrana activando (línea negra continua) a proteínas quinasas (ej. IKK y JNK). Estas proteínas inhiben a IRS1 (líneas punteadas) reduciendo la señalización insulínica. Asimismo, las especies reactivas del oxígeno (EROs) y el estrés del retículo endoplásmico (ER) estimulan a IKK y JNK. Lípidos específicos también interfieren en la señalización insulínica al activar la proteína kinasa C (por diglicéridos), la cual inhibe a IRS1, o reducir la actividad de Akt (por ceramidas).²
Resistencia insulínica mediada por lípidos
La RI está comúnmente asociada a desórdenes del metabolismo lipídico que incluye la acumulación tisular ectópica de lípidos, entre ellos en el músculo esquelético.³
Existen dos aspectos que requieren ser discutidos para una mejor comprensión de la relación entre los lípidos y la RI. Por una parte, identificar cómo los lípidos se acumulan en tejidos ectópicos. Por otra, cómo y cuáles son las especies lipídicas que inducen RI.
Sobre el primer punto, es claro que debe existir un desequilibrio entre la captación y oxidación de ácidos grasos que permita su acumulación en células de tejidos específicos. Por otra parte, individuos con RI tienden a caracterizarse por una menor densidad mitocondrial y síntesis de ATP en músculo esquelético. Basado en esta evidencia, algunos autores han propuesto la existencia de una disfunción mitocondrial en músculo esquelético de individuos con RI, lo cual determinaría una menor capacidad oxidativa de ácidos grasos y, en consecuencia, su acumulación intracelular.² Sin embargo, esta hipótesis ha sido ampliamente cuestionada, dado que una menor densidad mitocondrial no necesariamente determinará una menor oxidación de lípidos.¹
El segundo aspecto está referido a cómo los lípidos interfieren en la señal insulínica. Esto conduce a la pregunta de cuál o cuales especies lipídicas ejercen dicho efecto. Dado que los triglicéridos acumulados en músculo esquelético poseen una actividad biológica neutra, es decir, no interfieren en la actividad de proteínas, otras especies lipídicas debieran dar cuenta del efecto deletéreo sobre la señal insulínica. En este sentido, los diglicéridos o ceramidas han mostrado estar aumentados en músculo esquelético de sujetos con RI .¹
Respecto a cómo los lípidos ejercen su acción inhibitoria sobre la señal insulínica, la evidencia disponible es más concluyente. Se ha demostrado que los diglicéridos son capaces de influenciar la actividad de proteínas específicas, entre ellas, la proteína quinasa C. Esta proteína posee actividad serín-quinasa, es decir, fosforila a proteínas blanco en residuos de serina. Uno de los sustratos para la acción de proteína quinasa C sobre el IRS1, lo cual determina una atenuación de la actividad de la señal insulínica, de manera análoga a lo que ocurre en una condición proinflamatoria (Figura 1).²
Tejido adiposo: órgano central en la inflamación y resistencia insulínica
El principal tipo celular que compone el tejido adiposo (TA) es el adipocito, célula capaz de almacenar triglicéridos (TG) en su citoplasma sin ver afectada su fisiología. El tamaño de la gota lipídica del citoplasma está regulado por múltiples mecanismos, que en general incluyen la lipogénesis (formación de TG) y lipólisis (degradación de TG con salida de ácidos grasos libres a la circulación). En condiciones de balance energético positivo crónico, esta célula puede expandir su volumen hasta 1000 veces. El adipocito hipertrófico tiene una mayor tasa lipolítica, lo cual condiciona una mayor liberación de ácidos grasos no esterificados a la circulación, por lo tanto, mayor riesgo de acumulación ectópica de lípidos. Por otra parte, los adipocitos de gran tamaño poseen una mayor síntesis y liberación de productos de secreción del tejido adiposo (adipoquinas) que pueden deteriorar el metabolismo lipídico y glucídico, tener efectos pro-inflamatorios o pro-trombóticos, además de inhibir la diferenciación de pre-adipocitos en adipocitos. Existen múltiples adipoquinas de efectos delétereos cuya secreción está aumentada en los adipocitos hipertróficos, entre las que destacan la leptina, resistina, angiotensina, citoquinas pro-inflamatorias y quemoquinas. Paralelamente, estos adipocitos de gran tamaño secretan menor cantidad de adiponectina, una de las pocas adipoquinas con efectos antagónicos a los recién descritos. Producto de este ambiente auto/paracrino pro-inflamatorio, el TA es infiltrado por macrófagos, que a su vez secretan moléculas pro-inflamatorias, alterando aún más el perfil secretor del TA, lo que perpetúa el fenómeno.³


Figura 2. Tejido adiposo: órgano central en la inflamación y resistencia insulínica.
Todo lo anterior permite plantear que las características de la expansión de la masa adiposa influirán de forma importante en el desarrollo de alteraciones metabólicas propias de la obesidad, entre ellas la RI y la Diabetes Mellitus tipo 2.
Incluso un metaanálisis a partir de doce estudios homogéneos indican que los sujetos obesos tienen un riesgo de padecer diabetes tipo 2 siete veces superior al de la población general, mientras que en las personas con sobrepeso, la probabilidad es tres veces mayor. Estos resultados resaltan la importancia del manejo del peso corporal en la prevención de Diabetes tipo. ² ̓ ⁴
Referencias
1. Perez M., Gomez G. Obesidad, Adipogénesis y Resistencia a la insulina. DOI: 10.1016/j.endonu.2011.05.008
2. Carrasco N., et al. Síndrome de resistencia a la insulina. Estudio y Manejo. Rev Med Clin. Vol. 24. Núm. 5. Páginas 827-837. Septiembre 2013.
3. Pollak F., et al Resistencia a la Insulina: Verdades y Controverdías. Rev Med Clin. Volume 27, Issue 2, March 2016, Pages 171-178
4. Abdullah A., et al. The magnitude of association between overweight and obesity and the risk of diabetes: a meta-analysis of prospective cohort studies. Diabetes Research and Clinical Practice 2010; 89(3): 309-19.